2021.11.10
- Announcements
Unique Three-dimensional Structure and Functional Correlation of a Huge Proteolytic Enzyme Complex
- Experiment at Kibo
Successful results obtained from JAXA Protein Crystal Growth Project
Background and interest of the research
Proteins are responsible for various physiological functions in the body of humans, animals, plants, and microorganisms in common. There is a great deal of interest in what kinds of proteins exist and how individual proteins work, and scientists are working on them according to their own specialties. Among them, we are exploring the relationship between the three-dimensional structure of proteins and their physiological functions. For example, how oxygen is transported through the body after we breathe in, how nutrients are utilized after we eat, and so on. In order for an organism to function properly, these various functions must work properly. If any one of them is missing, it can cause illnesses such as difficulty in breathing or inability to exercise due to lack of nutrition. However, even these important proteins do not have an eternal life, and their functions gradually deteriorate as we age. A certain amount of repair takes place, but eventually they become unwanted proteins, which must be promptly removed from the body. To put it another way, if you leave them as they are, the body will turn into a pile of garbage, making it impossible to live. Among the proteins that have various functions, there are also proteins that have the ability to break down and remove those unwanted proteins. These proteins are named proteasomes, and they are enzymes with parts that are similar to other proteolytic enzymes (such as the proteolytic enzymes that digest meat protein after eating it) and parts that are completely different.
Proteasome was discovered and named in the 1980s as a very large complex (made up of many different proteins) that breaks down proteins in the cell. Normally, a protein is a globular molecule with a folded peptide chain that has a place where the protein performs its unique function (active site). The proteasome, on the other hand, is made up of 28 subunits (individual proteins), whose active sites are scattered throughout the complex. These 28 subunits are not completely different, but have almost the same three-dimensional structure (14 in eukaryotic cells and 2 in prokaryotic cells). The common structure of the system is roughly shown in Figure 1.
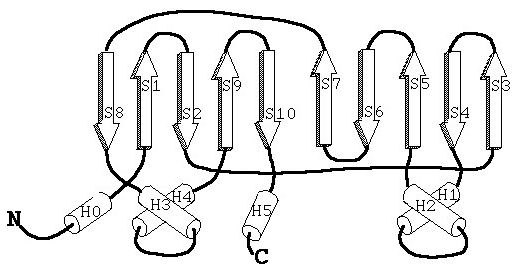
Starting from the N-terminus, the β-strand folds together to form an inverted parallel β-sheet, which is connected by several α-helices. Seven subunits come together to form a ring (this combination of ‘seven’ subunits is quite unique among biological substances), which is called the α- or β-ring. These rings stack on top of each other in an α-β-β-α pattern, resulting in the formation of a large barrel shape with cavities, as shown in Figure 2 when viewed from the side.
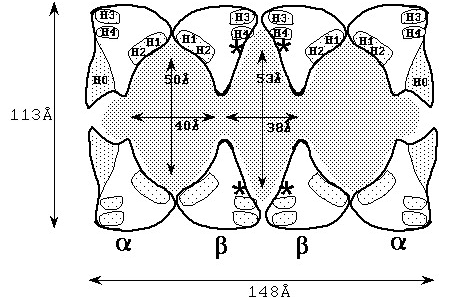
The white areas are the subunits, the gray areas are the cavities. The active sites are marked with asterisks (*), and the helices are marked with letters of H.Starting with the left ring, the pattern is α-β-β-α ring.
This figure shows a cross-sectional view from the side, and you can see that there are large cavities in the center and at both ends. When this enzyme was first discovered, it was not expected to have such cavities, and it was not possible to obtain the high-definition electron microscopy images that we have today, so it was a very interesting subject to study structurally.
This proteasome complex is called the 20S proteasome because its sedimentation coefficient, which indicates the size of the molecule, is 20S. It has active sites for protein degradation in the structure shown in Figure 1 (white areas in Figure 2), but as you can imagine from Figure 2, many active sites are scattered throughout the complex with some positional symmetry. At the same time, we can expect each active site to function in conjunction and cooperation with the other. It was revealed that the 20S proteasome not only degrades proteins, but also has the ability to cut out peptides that can present antigens for immunity. This function is characteristic of the mammalian proteasome, which has an immune mechanism. It was also found that stopping this degrading activity induces cell death (apoptosis) (cells are on the verge of bursting with unwanted proteins and cell functions stop). We have become very interested in where this kind of immune response function comes from in the three-dimensional structure. Furthermore, around the beginning of 2000, it was discovered that the proteasome was highly expressed in cancer cells (to cope with the large amount of immunoglobulins expressed to attack cancer cells), and that by stopping its degrading activity, the growth of cancer cells could be suppressed. As a result, it was proposed that inhibitors of proteasome activity could be used as anticancer agents. With such background, we have been strongly interested in the structural features and functional expression of the proteasome, and at the same time, we have been studied on three-dimensional structural analysis with the aim of developing effective anticancer drugs.
Results and contribution points of space experiments
As mentioned earlier, this proteasome size is 20S, and in terms of molecular weight, it consists of 28 subunits of about 30,000 Daltons, for an overall molecular weight of 750,000 Daltons. The X-ray structure analysis of such a large protein presents various difficulties that are different from those encountered in ordinary protein crystallographic analysis and crystallization. One of the drawbacks is that the crystal lattice is also large due to the large molecular size, making it impossible to collect a dataset with a laboratory X-ray diffractometer. Another drawback is that it is difficult to obtain good quality and stable crystals due to the large lattice. What does a good quality crystal look like in this context? One of the requirements is that the crystals should be clear, clean, without cracks, and have well-defined corners and edge lines. In addition, from a diffraction point of view, a good crystal is one that provides high resolution (ability to reflect X-rays). High resolution means ability to clearly separate and distinguish even small points.
In protein crystallization, the target protein, which has been purified to high purity, is mixed with a precipitant or other agent for crystallization, and the protein molecules are transformed from the liquid phase to the solid phase by changing the protein solution concentration ratio (crystallization). Crystallization is affected by a variety of physicochemical factors such as temperature, concentration, pH, ionic strength, and type of solute. The determination of these conditions is to some extent based on experience and various screening methods through trial and error. However, there is one condition that we just cannot suppress or control. That is gravity. For an average protein (molecular weight of a few tens of thousands), the effect of gravity might be negligible, as it is offset by free diffusion in the solution. However, gravity still has an effect on the local concentration gradient in the solution. In particular, convection in the solution will be greatly affected. If crystallization can be performed with reduced or no gravity, at least convection in the solution will be suppressed, and crystallization will proceed solely based on the interaction between the protein and the solute.
Based on this idea, we conducted proteasome crystallization in the Japanese Experiment Module "Kibo" of the International Space Station. Figure 3 shows the micrographs of the crystals obtained on the ground and in space.

Crystals of appropriate size were obtained for both ground and space. You can see that the crystals formed in space look slightly thicker, and the crystals formed on the ground have a little precipitation. The appearance under microscope does not tell us much more than this, and resolution and other factors cannot be determined without exposed with X-ray. In addition, we will discuss later the quality of the crystals, which will never come out from microscopic observation.
The above two types of crystals were subjected to diffraction experiments at SPring-8, a large synchrotron radiation facility. The maximum resolutions of the crystals were 2.24 Å (ground) and 1.93 Å (space), respectively, with the space crystal having the higher resolution. In terms of data completeness, the analytical resolutions were 2.92 Å and 2.74 Å, respectively. Here, we evaluated the physical quantity of crystal mosaicity when processing diffraction data (Figure 4). Mosaicity is the amount of misalignment between blocks (think individual lattices in a crystal as blocks) of nearly perfect microcrystalline regions in a crystal visible to the naked eye. This value can never be observed with the naked eye.
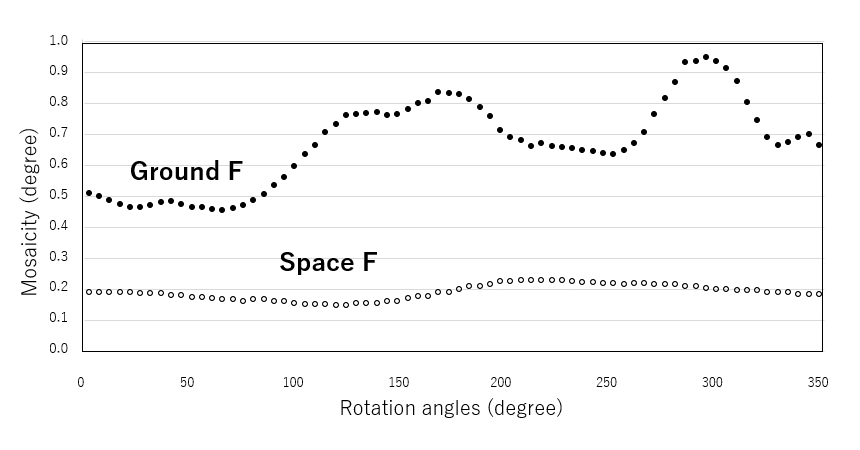
From this plot, it can be seen that crystals formed on the ground have a period of about 180 degrees per rotation, and the mosaicity value goes up and down between 0.5° and almost 0.8°, while crystals formed in space have almost no vertical variation, about 0.2° per rotation. Variations in crystals formed on the ground indicate that diffraction spots from crystals exposed to X-rays have higher mosaicity in certain directions, meaning that there is a larger displacement of micro perfect crystals (blocks) in certain axial directions of the 3D crystal. On the other hand, this is not the case for crystals formed in space, indicating that the misalignment is within a minimum range in all three axes of the crystal. This means that there is a high possibility that the crystals can be stacked stably in all three axes. This means that large, high-quality crystals can be obtained.
As a result of the analysis from the proteasome crystals obtained in this way, we were able to obtain the binding of the inhibitor (a specific drug for malignant myeloma) to the active site as shown in Figure 5, with an accuracy that allows us to interpret the interaction of each atom with the amino acid.
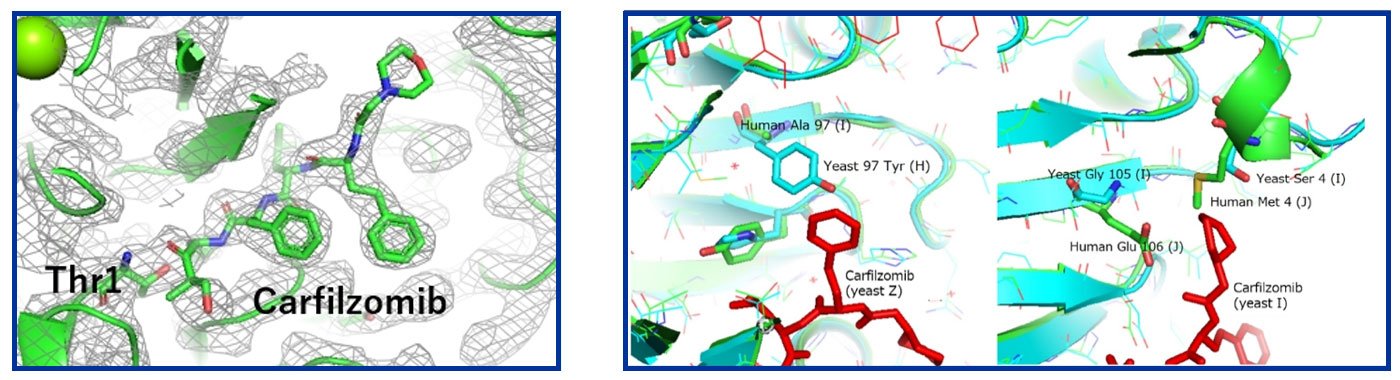
The left side of Figure 5 clearly shows the electron density of the inhibitor molecule in the crystals formed in space, while in the crystals formed on the ground, the resolution was low, and this location "appeared" to be the location of a water molecule or precipitant (MPD).
In a similar vein, we are currently analyzing inhibitor binding to the proteasome of human erythrocytes. Although the structure of the inhibitor-bound form is not yet available, as shown in the right side of Figure 5, a comparison of the structure with that of the yeast proteasome (blue) shows that the contact (green) at the inhibitor-binding site (the aromatic ring site opposite to the threonine side of the inhibitor) contains amino acids that are different from those in yeast, suggesting that the binding mode in humans may be different.
Future research
The relationship between the active site of the proteasome and the inhibitor described so far is for only one location inside the 20S proteasome molecule. As mentioned at the beginning of this article, the 20S proteasome is an aggregate of 28 subunits, and its active site has six functional locations corresponding to those described above. In addition, at least three types of active sites have been proposed, suggesting that the intramolecular degradation mechanism itself consists of a very complex interlocking structure of active sites. Through this analysis, we were able to follow the time series of inhibitor introduction and capture the binding to the six active sites in the molecule at different times. Instead of bonding at exactly the same time, some of them bond and then bond to other parts of the active site. In addition, we are now able to prove the inhibitory effect of blocking the entry pathway of the substrate (to be degraded), not necessarily the active site. It is considered that the interlocking, uncoordinated, and non-specific binding of these active mechanisms leads to the suppression of side effects of anticancer drugs. Therefore, it is essential to analyze not only the local bonding mode, but also the overall structure from a bird's eye view, and we would like to proceed with this analysis through creating high-quality crystals in space.
Researcher

MORIMOTO Yukio
Professor
Biomolecular Structure Laboratory, Division of Radiation Life Science,
Institute for Integrated Radiation and Nuclear Science, Kyoto University

Related News
-
 2025.11.13
2025.11.13- Announcements
The Asian Try Zero-G 2025 Flight Items were Launched!- Experiment at Kibo
- Kibo Utilization Office for Asia (KUOA)
-
 2025.10.14
2025.10.14- Announcements
Three CubeSats successfully deployed from "Kibo"!- Experiment at Kibo
-
 2025.10.01
2025.10.01- Announcements
The preliminary rounds for the 6th Kibo Robot Programming Challenge were held in each participating country and region!- Experiment at Kibo
- Kibo Utilization Office for Asia (KUOA)
-
 2025.09.29
2025.09.29- Announcements
3 CubeSats will be deployed from “Kibo” Japanese Experiment Module on Friday, October 10th, 2025.- Experiment at Kibo
-
 2025.09.22
2025.09.22- Announcements
Five CubeSats successfully deployed from "Kibo"!- Experiment at Kibo
Unless specified otherwise, rights to all images belong to ©JAXA