![]()
![]()
02

Most proteins in living organism on Earth are composed of L-amino acid. D-amino acid has long been believed to be only present on the cellular barriers of certain bacteria. But recently, D-amino acid was proved to be present in bodies of mammal as well. Besides, it was reported that D-aspartic acid exists in human lesional tissues affected by diseases (e.g. Alzheimer's disease and cataract) triggered by abnormal protein folding and aged dermal tissues. Few reports have been published about peptidase that specifically recognizes D-amino acid. The D-aspartyl endopeptidase (PAE) in particular is the only example of enzyme derived from bacterium.
To date, we synthesized a new D-aspartic acid substrate and separated a D-aspartyl endopeptidase producing microorganism (Paenibacillus sp. B38 strain). We named the producing enzyme "paenidase" or "PAE" and analyzed its nature (S. Takahashi et al., J Biochem, 2006). We then identified the genes of PAE, constructed a massively expressing system and estimated PAE's active sites in an amino acid variation method. Furthermore, we published a method for analyzing peptides degraded with PAE (H. Maeda et al., Anal Chem, 2015) and reported that lesional tissues affected by a chronic obstructive lung disease contain D-aspartic acid (M. Ogasawara et al., Exp Lung Res, 2016). In these reports, we showed that PAE was extremely useful as a tool for detecting D-aspartic acid.
While knowledge of cell biology and biochemistry has thus accumulated, the structural basis of PAE activity has never been clarified. PAE's mechanism of recognizing D-aspartic acid long remained unknown to us deprived of comparable reference objects. In fact, we searched for substrate recognition sites, using data on sequence homology with similar enzymes and conducting a homology modeling, but we never succeed in identifying the substrate recognition sites. Then, we thought that the mechanism of presenting enzyme activity would be identified by determining the precise 3D structures of active sites and substrate recognition sites through crystal structure analyses of PAE. Thus, we once attempted crystal structure analyses in a separate joint research, but these structures were not determined.
The research activities for structural analyses of PAE were interrupted for a while. And at the 2016 Annual Meeting of the Japan Society for Bioscience, Biotechnology and Agrochemistry, held on March 27 to 31, 2016, in Sapporo, Japan, we fortunately had a talk with some researchers of JAXA, which resulted in our participation in JAXA High-quality Protein Crystal Growth Experiment. In those times, no member of our team was specialized in X-ray crystal structure analysis. Therefore, the roles were divided. JAXA would assume the search for crystallization conditions and structural analyses and our team the provision of protein samples, substrates and an inhibitor.
Several months after the beginning of the joint research, new crystallization conditions were determined. In February 2017, we conducted the first space experiment "LTPCG#1" during which high-quality crystals of resolutions of up to 2.25 A were successfully obtained. We were the first in the world to identify the structure of "D-aspartyl endopeptidase (PAE)" (Fig. 1). With crystals obtained on Earth, the highest resolution was about 3.5 A. The improvement of crystal qualities during the space experiment contributed to the more precise structural analysis of proteins. PAE crystals produced during the space experiment were soaked with the D-aspartyl endopeptidase inhibitor "i-DAEP (Bz-Arg-His-[D-Asp]-CH2Cl). Co-crystals of the Enzyme and inhibitor complex were obtained. Analyses of the co-crystals (Fig. 2A) revealed that the side chain of D-Asp residue of i-DAEP was bound with the side chain of PAE (Fig. 2B).
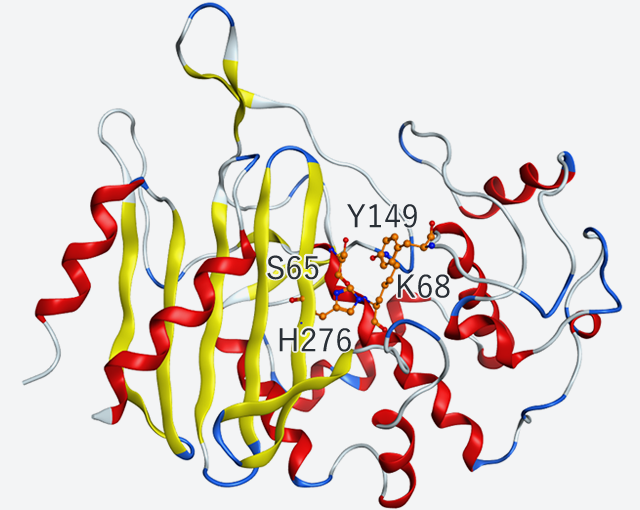
Fig. 1
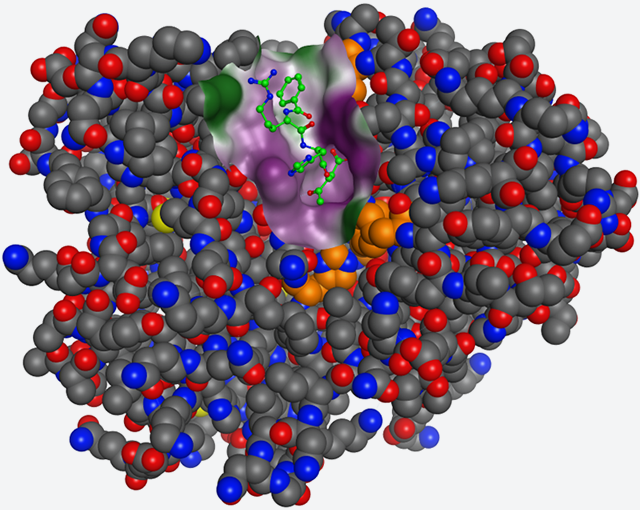
Fig. 2(A)
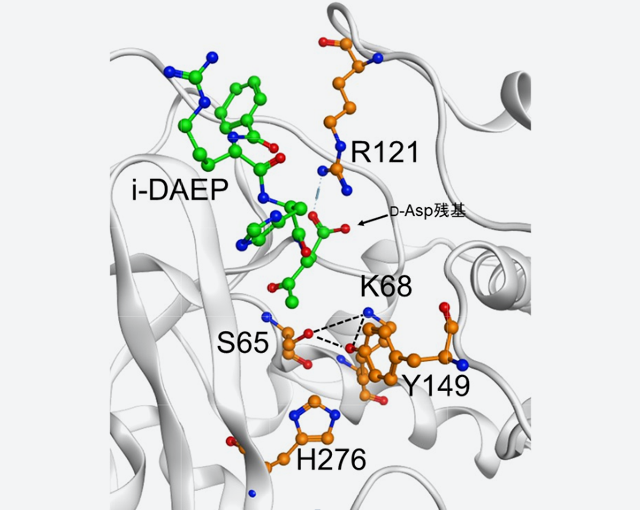
Fig. 2(B)
Fig. 1: Tertiary structure and active residues of D-aspartyl endopeptidase (PAE)
Fig. 2: Structure of a complex of D-aspartyl endopeptidase (PAE) and inhibitor i-DAEP
(A) Tertiary structure of a complex; i-DAEP is represented in green and Bz groups and Cl atoms are not indicated.
(B) The side chain of the 121st Arg residue of PAE (R121) interacts with the side chain of the D-Asp residue of i-DAEP.
Since we were aware in the preceding research activities that PAE had a nature similar to that of the group of cell wall synthetic enzymes from bacteria, we superimposed for comparison the structure of PAE on the structure of the group of cell wall synthetic enzymes (Fig. 3). In both structures, the enzyme active sites Ser, Lys, Tyr and His were in accord and the structures of the loop containing Arg121, PAE's substrate recognition site, greatly varies with the enzyme. Thus, we concluded that the difference of these enzymes' substrate specificities was attributed to the type and steric conformation of the amino acid residues contained in the loop.
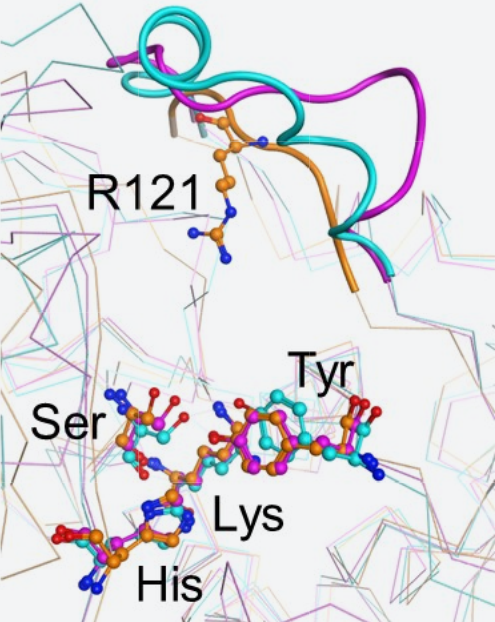
Fig. 3
Fig. 3: Structural superimposition of D-aspartyl endopeptidase (PAE) and group of cell wall synthesized enzymes
Orange: PAE
Blue: D-Ala-D-Ala carboxypeptidase B (derived from Streptomyces strain R61)
Pink: Alkaline D-peptidase (derived from Bacillus cereus)
Ser, Lys, Tyr and His represent respective active sites; thick line: loop region.
Next, the substrate recognition site Arg121 was replaced to different amino acids (Ala, Ile, Glu, His and Lys) and the enzyme activities of R121A, R121I, R121E, R121H and R121K were analyzed. The replacement by Lys (R121K), a basic amino acid like Arg, enabled forty-four percent of the activity to be retained (Fig. 4). The replacement by Ala, Ile, Glu and His resulted in considerable decrease in activity (Fig. 4).
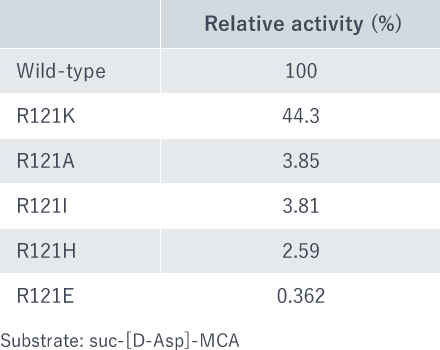
Fig. 4
Fig. 4: Relative activities of mutant enzymes. As substrate, suc-[D-Asp]-MCA was used.
The analyses showed that positive charges of the side chain play an important role in substrate binding. Furthermore, the enzyme activity of these mutants were analyzed using the peptide from the N-terminal to the tenth residue of amyloid beta protein (Aβ). In this peptide, the seventh Asp residue is replaced by D-Asp. With wild-type, the C-terminal side of the seventh D-Asp residue was cleaved and completely degraded to two peptides. Interestingly, peptides were not completely resolved with the mutant enzymes R121K or R121A. When the peptide is a substrate, the steric conformation of the side chain (Arg or Lys) may greatly affect the reaction kinetics of the enzyme.
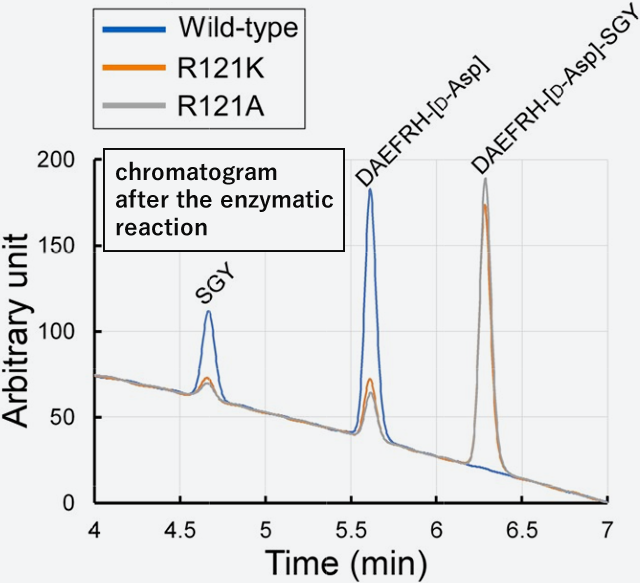
Fig. 5
Fig. 5: Degradation of D-type Asp-Containing Aβ Decapeptide by Various Mutant
A high-performance liquid chromatography was applied after enzyme reaction. Wild-type (blue line) is completely degraded to 2 peptides (DAEFRH- [D-Asp], and SGY), whereas the mutant enzyme (Orange and grey lines) is poorly degraded.
Our understanding of PAE's substrate recognition mechanism initially made a slow start but made a tremendous advance after the recent 3D structure analyses. We hope that we will utilize the new findings to search for new antibacterial agents and analyze their inhibiting mechanism and apply them to development of disease diagnoses (Alzheimer's disease and cataract) based on detection of D-aspartic acids.

NIRASAWA Satoru, Senior Researcher