![]()
![]()
![]()
![]()
About
03

About01.
What is protein?
About02.
Why is the study of proteins important?
About03.
Protein structure determination
About04.
Proteins around us
Protein molecules are too small to observe directly,
so studying them is always accompanied by some difficulty.
We will show you how we work on protein molecules.
A protein is made up of tens to thousands of amino acids and because it is only up to a few tens of nanometers (nm) in length (1 m = 1,000,000,000 nm), it is too small for the naked eye to see. If a protein molecule assumed the relative size of a human, it would be the size of Jupiter, or about 140,000 km in diameter.
Even with the latest microscope, we cannot see the structure of such a tiny molecule in detail. Instead, we use crystals and X-rays. Crystals are solids in which atoms and molecules are repeatedly arranged. You might think of salts and minerals when it comes to crystals, but complex molecules such as proteins consisting of many amino acids can also be crystallized (Fig. 1).
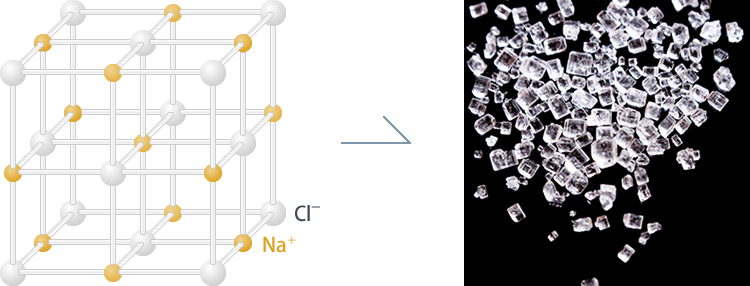
Fig. 1a Salt crystals (sodium chloride)
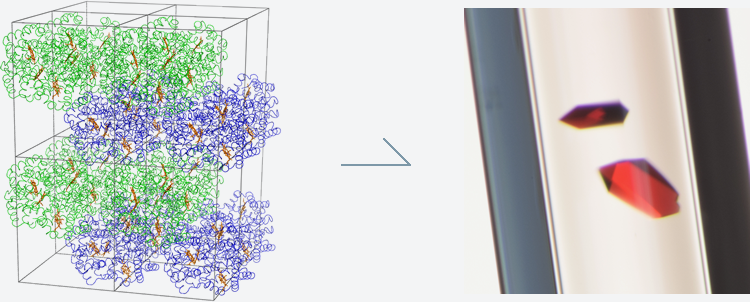
Fig. 1b Protein crystals (hemoglobin)
X-rays are a type of electromagnetic wave. For now, just consider it a wave with a certain wavelength. When an X-ray hits a crystal, it is only reflected in certain directions. The reflected X-rays can be observed as spots on a detector placed behind the crystal (Fig. 2). This process is actually diffraction, but we often use the term reflection because it could be interpreted that a crystal reflects an X-ray.
Appearing like many random spots on the screen, the crystal's structural information is packed in this image. More precisely, the position and intensity of individual spots contain information on the repeating pattern and protein structure in the crystal. The spots are spread in a three-dimensional space, so we shoot a crystal with an X-ray beam from various directions to measure the positions and intensities of individual spots. The collected data are processed and analyzed with special programs, allowing us to calculate the crystal's molecular structure. This technique is called X-ray crystallographic structure determination.

Fig. 2 An example image of diffraction spots
Crystal quality is crucial when determining a protein structure by using crystals and X-rays. The higher the crystal quality, the more detailed the visible structure.
What's wrong with a poor crystal? The theory of X-ray crystallography was established based on two findings: the diffraction of X-rays discovered by German physicist Max von Laue in 1912, and Bragg's law proposed by British physicists William Henry Bragg and William Lawrence Bragg (father and son). You might not be familiar with the theory. It is often taught in the last year of high school and university. If you never heard of it, remember when you played with a long skipping rope and then look at Fig. 3. Here, we only give a brief explanation, but you can access advanced information by using such keywords as "X-ray diffraction," "Bragg's law" and "X-ray crystallography."
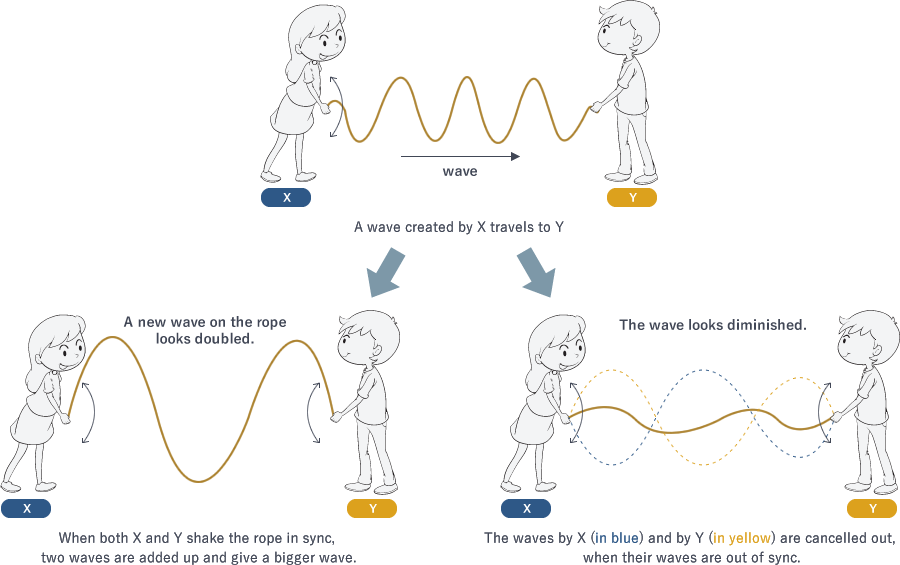
Fig. 3 Adding up waves
For a high-quality crystal in which molecules are well ordered, the incoming X-ray waves are diffracted in certain directions. When the peaks and troughs of the outgoing waves overlap, they are added up to create a bigger wave. When there are many well-ordered protein molecules in a crystal, diffracted waves are strengthened and give thick and sharp spots on the screen.
On the other hand, diffraction by a poor crystal yields blurred spots because the waves from disordered molecules in the crystal are oriented in slightly different directions and make spread-out spots. In the worst case, spots overlap the adjacent ones and can no longer be detected (Fig. 4).
Because the position and intensity of individual spots contain information about the protein structure in the crystal, measuring precise and accurate diffraction data is crucial for determining the structure; in other words, inaccurate diffraction data only provide an inaccurate structure.
Imagine that you want to determine a structure of a disease-causing protein. Although you manage to crystallize it, the quality is poor. From such a low-quality crystal, you cannot obtain accurate diffraction data and the resulting structure will also be of low quality. How could you rationally design a medicine to treat the disease or evaluate the risk of side effects, even if you find an effective molecule? Without knowing how to do so, such a medicine may not be safe to use.
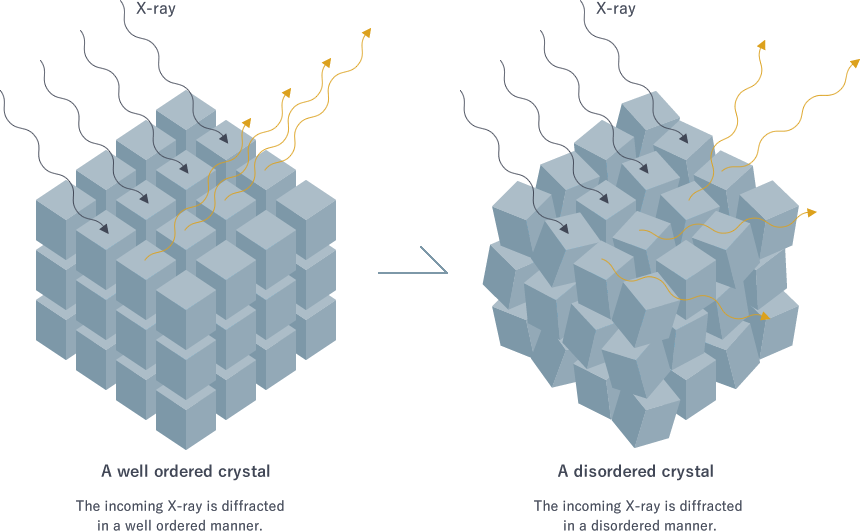
Fig. 4 Crystal quality affecting diffraction spots
It is extremely difficult to grow high-quality protein crystals. Searching for good crystallization conditions often takes years, and in some extreme cases, crystals cannot be improved enough to give a detailed structure after more than 10 years of optimization. The technique of protein structure determination has been hugely advanced thanks to improved computer performance and the availability of large synchrotron facilities like Photon Factory and SPring-8 in Japan(*1) as intense X-ray sources. In contrast, crystal growth and optimization, which are among the most critical steps for structural studies, still rely on skills and efforts on a trial-and-error basis by individual researchers.
What makes protein crystallization difficult? One reason is the convection generated by the earth's gravity. A crystal grows by assembling molecules around a nucleus formed in the solution (Fig 5). The slower the crystals grow, the better their quality. However, convection accelerates crystal growth and makes crystals less ordered as the molecules accumulate around the nucleus in a hasty manner. Don't you think that you could tidy up your room better by doing it slowly at your own pace, instead of being forcibly rushed by someone else?
(*1) Synchrotron X-ray is a converged intense electromagnetic wave. It is generated by accelerating electrons at nearly the speed of light and then passing the electrons through special magnets.
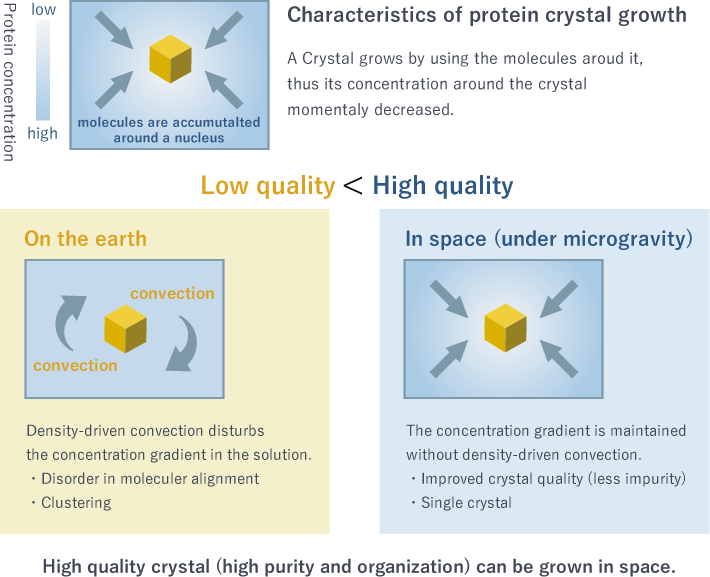
Fig. 5 Crystal growth in space
The International Space Station can offer the solution. The convection on the earth is a long lasting headache for scientists. One of distinctive features of space is a gravity-free environment, where no convection occurs due to density and/or temperature differences. Under conditions that do not exist on the earth, protein molecules can be gathered in an ordered manner and you have a better chance of determining the structure by obtaining high-quality crystals that cannot be grown on the earth.
Our main objective is to contribute to wide-ranging research and development by offering a microgravity environment where better crystals can be grown.

If you would like to know more about crystallization in space, follow the link below.
Protein Crystal Growth on the International Space Station using the Japanese Experimental Module ("Kibo")